- 无参组装原理及组装方式的选择
1、组装软件:Trinity(软件相关文章)
2、reads 双端分析 150bp
图1 组装图
图2 具体组装过程
无Contig数据,提供转录本数据、Unigene数据
3、文件名:Final-Transcript、Final-Unigene
Final-Transcript转录本文件:Fasta文件——EditPlus软件打开
转录本命名方式:c49888.graph_c0_seq1
c49888.graph:编号
c0:Component (片段集合)
c49888.graph_c0 类似于基因编号
seq1:前缀一样 seq不一样 指不同转录本来自同一个Unigene基因同一个Component
Final-Unigene:Fasta文件——EditPlus软件打开
一个基因有多个转录本,外显子差异,通常选择最长转录本序列代表Unigene基因序列
4、组装结果统计表格
注意Unigene基因数量多少,数量过多,拼接结果差(常见作物正常基因数量2-3万,拼接的话6-9万结果不错)
N50:计算方式,所有序列计算全长,N50越大说明拼接长片段多
5、无参/有参区别
a.无参适用范围广
b.软件分析依赖算法,存在误差错误,对于相似基因无法辨别装成一个基因。
c.基因表达量较低,会把较长基因装断,成俩个较短基因
d.无法判断是否污染,对分析结果影响大
6、组装结果不好,如何优化
如果200-300bp数据较多,其可靠性低,如果不进行定量,可以过滤掉。
定量后,表达量低,无read支持转录本,可丢掉。
7、组装方式:合并组装/分组组装
合并组装:同一个物种建议合并组装
分组组装:特殊需求,一个物种的两个亚种,一个植株的不同组织部位。只有最长转录本,对定量结果有一定影响。
两个物种做两个无参组装再进行比对。
- CDS预测
结果文件:BMK_5_Unigene_Structure——CDS
best:筛选出最好的蛋白序列,但不一定是最完整。文件夹中:
cds-序列文件:ORF:5prime_partial 无起始密码子/complete完整的/interal 中断的,既无起始密码子也无终止密码子/3prime_partial 无终止密码子
pep-翻译的蛋白序列
gff-关于结构的定义
基因编号是软件输入顺序
complete:在best中提取的 预测的CDS结果中有明确的起始密码子和终止密码子
Transdecoder软件预测
正向预测三次,每隔一个碱基预测一次,反向也是。
(NCBI可以提交转录本数据预测CDS)
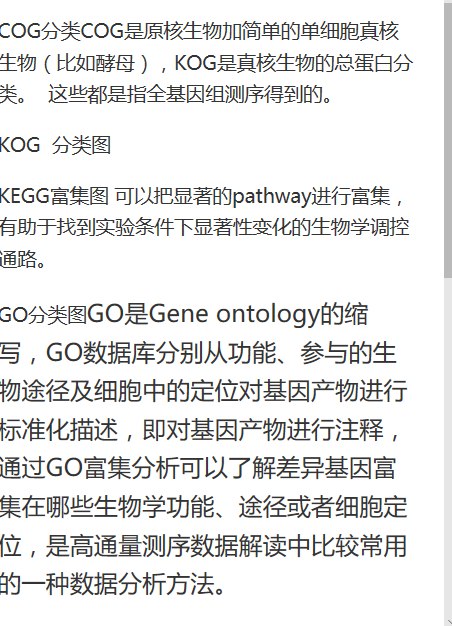
- Unigene功能注释
Blastx蛋白序列比对NR,Swissprot,COG,KOG,eggNOG,KEGG,Pfam
百迈克筛选条件:evalue<1e-5
blast2go软件:GO
参考NCBI数据关系,关联出GO结果
注释结果文件:BMK_4_Unigene_Anno
All_Database_annotation:筛选基因常用表格
NR注释:比对来源物种图
可以只注释一个物种:例如把蛋白序列只以金银忍冬注释
- 定量及相关性分析
定量软件:RSEM 针对转录本进行定量,累加到Unigene。
判断是否表达可参考文献设定标准。FKPM<1/5/10
样品间相关性评估:皮尔逊相关系数r^2>0.8 认为相关性比较好
- 差异分析及差异基因富集分析
差异分析软件:有生物学重复:DEASEQ2,DESeq,edgeR
筛选条件:FC(log2FC),FDR(PValue)
差异基因功能富集:GO,KEGG
- 基因检索方法
寻找目标基因 :根据已知的基因名,在功能注释中进行检索
差异分析结果中找关键基因:根据差异基因的功能富集结果(GO/KEGG),找关键基因。筛选q值 <0.01/0.05